
Abstract
This study provides an efficient alternative by extracting bioactive compounds from Haematococcus pluvialis via matrix solid-phase dispersion (MSPD) from its wet form, reducing one of the process steps with the greatest economic impact, the drying of the microalga. To obtain a suitable extract for nutraceutical purposes, solvents with the generally recognised as safe (GRAS) designation (ethanol, ethyl lactate, and ethyl acetate) with limitations of use (acetone) and extractants with higher toxicity such as methanol and methyl-tert-butyl ether (MTBE) are contrasted. Through the optimisation of the extractive process, ethanol, a GRAS solvent, presents the best overall recovery for carotenoid compounds and fatty acids, showing an antioxidant activity of 1.58 mmolTE g−1 DW, comparable to its synthetic alternative of petrochemical origin without the drawback of having limitations in its food use. In addition, the identification of the phenolic compounds, phloroglucinol, p-coumaric acid, gallic acid, and catechin, not previously characterised in red stage H. pluvialis, provides a response to the phenolic activity present in the extract (24.65 mmolGAE g−1 DW). Comparison of the extractive efficiency obtained with the main methods for the extraction of carotenoids and fatty acids in H. pluvialis, in contrast to the proposed method, shows a positive feasibility of this approach.
Similar content being viewed by others


Introduction
About 95% of the astaxanthin produced worldwide is the result of synthetic production (Jannel et al. 2020), which, although it has proven not to be as beneficial as its natural counterpart, its low manufacturing costs in relation to extraction from Haematococcus pluvialis have made it the preferred route to produce this carotenoid (Panis and Carreon 2016). Accordingly, the optimisation of extraction methods for maximum astaxanthin recovery from H. pluvialis has become one of the main objectives dealing with this microalga (Mularczyk et al. 2020). The current industrial process generally follows a two-stage approach, where green vegetative cells and astaxanthin-rich aplanospores are separately produced. The separation of both stages allows precise optimisation towards enhancing cell growth in the former and boosting astaxanthin synthesis in the latter. The use of optimised growth media that promote cell division in a harmonious environment is nowadays a common practice, whilst subjecting the culture to conditions that completely hinder or diminish cellular growth is used to trigger astaxanthin accumulation (Fábregas et al. 2000). Amongst the latter, nitrogen starvation and a high irradiance may be used in combination yielding high astaxanthin contents (Pereira and Otero. 2020). Once cultivation ends, H. pluvialis biomass may be recovered in the form of a de-watered concentrated paste by centrifugation. The microalgal paste may be then disrupted and dried, which improves final extraction efficiency (Lorenz and Cysewski. 2000).
The optimisation of the industrial production of astaxanthin has been mainly focused on improving carotenogenesis during the red stage, modifying the culture conditions to obtain a microalga loaded with as much astaxanthin as possible. In fact, over the last 30 years, studies based on this microalga have optimised the culture conditions to obtain a biomass with 2–6% (w/w) of astaxanthin, which seems to be the upper limit for this species (Do et al. 2016). Once the biological capacity of H. pluvialis to synthesise this carotenoid has been depleted, other approaches aim to increase astaxanthin production by modifying the last extractive step.
A limited extractive availability of this carotenoid astaxanthin contained in H. pluvialis, due to a strong, thick, and rigid cell wall present in its aplanospore stage, is a characteristic hindrance in this alga (Ye et al. 2020). Conventional techniques, such as Soxhlet extraction (Irshad et al. 2019a), have been used to penetrate the cell, but drawbacks that include long extraction times, high temperatures, and toxic solvents turn it into a costly alternative and difficult to apply in the nutraceutical field (Chen et al. 2020). Green solutions such as microwave-assisted extraction (MAE) has been used for the extraction of astaxanthin from H. pluvialis (Ruen-ngam et al. 2011). Although it has the advantage of working with wet samples, paradoxically it requires large volumes of solvent in relation to the sample (100:1 acetic ether) as well as temperatures above 40 °C, obtaining 0.8% DW as the highest average recovery, a low yield considering that astaxanthin is 6% of the cells (Kapoore et al. 2018). Similar problems are found with ultrasound-assisted extraction (UAE) or supercritical fluid extraction (SFE), in which the temperatures required for cell disruption generate significant effects on the stability of carotenoid compounds (Zhao et al. 2006). Thus, these approaches face a complex trade-off between disruption efficiency and energy consumption, resulting in complex scaling, such as the balance between optimal operating conditions and extract integrity (Kapoore et al. 2018).
To improve the extraction efficiency of traditional methods, it is indispensable to apply a preliminary drying of the biomass, which is one of the costliest steps in the extraction process (Shah et al. 2016). In highly sophisticated productions, spray drying or lyophilisation is used with barely any impact on the quality of the product (Stramarkou et al. 2017). Less sophisticated drying methods, such as solar drying, may reduce cost of this process step. However, such techniques may compromise the integrity of the carotenoid compound (Molina et al. 2003). There are currently no industrial methods that bypass these procedures due to the single focus on obtaining astaxanthin, which, for refining, requires the removal of moisture in the paste (Ahmed et al. 2015).
A pioneering proposal has focused on the extraction of wet H. pluvialis from the microalgae in its red stage using dimethylaminocyclohexane (DMCHA) as the extractive solvent, obtaining significant yields (Huang et al. 2018). In contrast, due to the toxicity of DMCHA, the extract generated, although promising, is incompatible with a nutraceutical purpose. On the other hand, different perspectives have been given to H. pluvialis understanding the microalga as a whole, thus assessing its quality not only for its content of this high-value carotenoid, but also as a source of fatty acids, carbohydrates, and proteins (Cerón et al. 2007). Therefore, a direct, scalable, and energy-efficient method to extract bioactive compounds from wet microalgal paste avoiding biomass pre-treatment or subsequent volatilisation of the extractant would be an attractive approach.
A simple technique directly applicable to semi-solid, solid, and viscous samples is matrix solid-phase dispersion. MSPD was first introduced by Barker et al. (1989) as a process for sample preparation, having the advantage of combining maceration, extraction, and filtration in a single process, eliminating centrifugation, drying, and separation steps. This extractive technique incorporates micro-milling through selected disruptive media capable of conferring disruption at the cellular level. In turn, it has a desiccant effect that provides homogeneous chromatographic packing, improving the interactions between the extractive mixture and the extracting solvent, creating a clean and efficient extract, outperforming conventional milling and extraction techniques. Based on these guidelines, Lores et al. (2016) developed and patented a scalable extractive approach, referred to as MSAT (Medium Scale Ambient Temperature systems) for obtaining bioactive extracts at pilot scale. Its applicability on an industrial scale was subsequently demonstrated (Gato et al. 2021; Rama et al. 2021) processing biomasses with a moisture content up to 90% without prior pre-treatment and obtaining high yields. The development of the technique and its applications faithfully comply with the principles of green chemistry (Anastas and Warner 1998).
The application of MSPD in the field of microalgae has been limited, being reported for the first time to obtain bioactive compounds in the marine species Isochrysis zhangjiangensis and Nannochloropsis oculata; using small amounts of solvent, recoveries, reproducibility, and extractions comparable to techniques such as ultrasound-assisted extraction were achieved (Jin et al. 2019). Meanwhile, the first application to freshwater microalgae was using H. pluvialis in its lyophilised state, efficiently obtaining a variety of bioactive compounds (Castillo et al. 2020). The response of this approach was evaluated by comparison with industrial disruptive systems, showing a 20% improvement in the recovery of the main carotenoid compounds astaxanthin, lutein, zeaxanthin, and β-carotene. At the same time, the addition of water to various extractants was proved to give better results in the recovery of amphipathic fatty chains of the omega-3 and omega-6 groups than pure solvents. In this way, the idea of using the original own moisture contained in the microalgae is attractive, eliminating the drying processes and its contradictory subsequent hydration.
Thus, the aim of this research is to obtain an extract rich in bioactive compounds from H. pluvialis red aplanospores, without the use of a previous drying step, starting directly from its raw wet form. The extractive efficiency is analysed by contrasting it with specific methodologies for obtaining fatty acids and carotenoids from lyophilised microalgae. Moreover, the antioxidant activity and polyphenolic content are evaluated, as well as the characterisation of the compounds responsible for these activities, to obtain the whole bioactive profile of the generated extract.
Materials and methods
Chemicals and reagents
The standards used for the identification and quantification of the main bioactive compounds contained in Haematococcus pluvialis (carotenoids, fatty acids, carbohydrates, and polyphenols) with their CAS numbers, purity, and suppliers are summarised in Supplementary Table S1. MS grade ultrapure water, ethanol, ethyl lactate, ethyl acetate, acetone, methanol, methyl-tert-butyl ether (MTBE), and dimethylformamide (DMF) were purchased from Scharlab (Barcelona, Spain).
Individual stock solutions of each carotenoid were prepared in DMF according to the supplier’s recommendations. Additional dilutions were prepared in ethanol for method calibration, characterisation, and quantification, as this solvent was used for the recovery assays. Apocarotenal, supplied by Dr. Ehrenstorfer (Augsburg, Germany), was used as internal standard. For the quantification of fatty acids and polyphenols, standards were prepared in ethanol, using nonadecanoic acid supplied by Tokyo Chemical Industry (Tokyo, Japan) and quercetin supplied by Sigma-Aldrich Chemie GmbH (SIGMA), respectively, as internal standard. For carbohydrate identification, standards were diluted in water and further diluted in ethanol. All solutions were stored in amber glass vials and protected from light at − 20 °C. All solvents and reagents were of analytical grade.
Red stage Haematococcus pluvialis biomass
Haematococcus pluvialis red aplanospore in paste form were recovered from an industrial scale production after 7 days of cultivation under inductive conditions of high irradiance and nitrate starvation. The optimised procurement procedure is described by Pereira and Otero (2020). In summary, H. pluvialis cultures were developed in photobioreactors following a two-stage approach. When the green vegetative culture reached the target density, the culture parameters were modified to favour the accumulation of astaxanthin. The astaxanthin-rich microalgal biomass (∼5% dry cell weight) was de-watered by centrifugation to a paste with about 25% solid content. Then, the paste was stored frozen at − 20 °C to avoid product degradation.
Matrix solid-phase dispersion
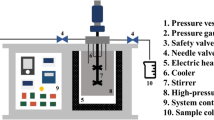
The wet H. pluvialis was directly weighed at the different study sizes, on a mortar. Then, SiO2 (50–70 mesh particle size) (Sigma-Aldrich, Germany) was added as a disrupting agent at the tested ratio, and it was disrupted for 10 min under red light. Disruption was carried out using a glazed porcelain mortar and pistil, to reduce the contact with the porous material and thus avoiding sample loss. A 20 μm PTFE frit was put at the bottom of a MSPD polypropylene cartridge (Fig. 1), as well as 1 g of dispersant. The disrupted microalgae mixture was then transferred to the cartridge and compacted with another PTFE frit on the top. Finally, elution was performed with 5 and 15 mL of the corresponding solvent, depending on the assay. The extraction time was maintained constant (20 min) by regulating the flow rate using discharge valve.

MSPD extractive procedure for wet Haematococcus pluvialis red aplanospore
Fatty acid recovery
The efficiency of MSPD for the extraction of saturated (SFA), monosaturated (MUFA), and polyunsaturated (PUFA) fatty acids was evaluated employing the Bligh and Dyer method (1959) modified for wall disruption of H. pluvialis. Briefly, 30 mg of lyophilised biomass was macerated in 2 mL of chloroform/methanol (1:2, v/v) and kept 24 h at 4 °C in the absence of light. The biomass was then disrupted using a homogeniser for 15 min, which incorporates a high-performance IKA T25 rotor–stator system at 24,000 rpm in an ice bath, preventing heat generation in the disruptive process. The supernatant was then decanted and stored. To ensure complete extraction, 5 mL of extraction mixture was added to the remaining solid and disrupted again for decanting and storage. Subsequently, 4 mL of Milli-Q water was added to each extract, and the contents were gently shaken to remove water-soluble impurities. The tubes were then centrifuged at 1800 × g for 10 min for bilayer separation. The lower lipid phase was carefully decanted from the upper residue and its moisture content removed by addition of 1 g sodium sulphate. For separation of the sulphate crystals, the extractive setup performed for MSPD was used, exchanging the filter medium for the sulphate crystals, passing the extract through it, performing a triple function: drying of the sample, separation of the desiccant and the extract, and filtering through a 20-μm PFTE membrane. The lipid extract solvent was volatilised under an extraction hood. These dried lipids were then measured gravimetrically.
Carotenoid recovery
To analyse the extractive efficiency of MSPD for the recovery of the main carotenoids in H. pluvialis, it was compared with the method developed by Kim et al. (2013) for the extraction of carotenoids from Dunaliella tertiolecta. Due to the difference in cell wall hardness between H. pluvialis and D. tertiolecta, the disruptive step was modified by adding mechanical disruption. Initially, 10 mg of lyophilised H. pluvialis sample was extracted with 1 mL of acetone and vortexed for 20 s. Cell wall disruption was carried out in the same way as in the lipid recovery process. The extract was then centrifuged at 1800 × g for 10 min. The extraction was repeated four times until the extract became colourless. The supernatant was then collected and filtered through a 0.22-μm PTFE membrane. A 0.7 mL aliquot was removed from the final extract and mixed with 0.2 mL of MiliQ water and 0.7 mL of hexane containing 0.1% w/w BHT. After gentle mixing, the top layer of hexane (0.5 mL) was separated and reconstituted in 0.5 mL ethanol. The procedure was developed in triplicate in a dry environment under red light to avoid possible degradation of the extracts.
UHPLC-QToF analysis (untargeted characterisation and fatty acids, carbohydrates, and polyphenols quantification)
For the initial analysis of carotenoid compounds, fatty acids, and carbohydrates, the obtained extracts were stored at − 20 °C in their respective solvents, adding 0.1% w /w BHT to avoid degradation. In turn, for the targeted quantification of the dried lipids obtained from the recovery study, the extracts were volatilised under a N2 stream and reconstituted in 500 μL of ethanol. For the characterisation and subsequent quantification of the polyphenols contained in the ethanolic extract of H. pluvialis via MSPD, and for all extracts analysed via UHPLC-QTOF, a previous filtration through 0.22-μm PTFE filters was performed.
Calibration standards for fatty acids and carbohydrates were prepared in ethanol covering a concentration range with six levels (0.1; 0.3; 0.5; 1; 3; 5 mg L−1). For the polyphenolic study, lower concentration ranges were injected, with 8 levels (0.01; 0.03; 0.05; 0.1; 0.3; 0.5; 1; 3 mg L−1). Each level was injected at least three times. The method showed a directly proportional relationship between the concentration of each compound and the chromatographic response, obtaining coefficients of determination (R2) higher than 0.992 in all cases, (Supplementary Figure S1). The quantification of the real samples was carried out by external calibration using stock solutions of the compounds studied in ethanol. The corresponding internal standards were added to the calibration standards and samples to correct for possible analytical variability. In all cases, the chromatographic response was equivalent in the spiked samples as well as in the real samples and in the calibration curve. Therefore, it was used as an additional measure to control the procedure, but no correction of the internal standard (IS) was necessary.
Based on the chromatographic protocol described by Otero et al. (2017) focused on lipid quantification of H. pluvialis, modifications were made for the combined untargeted characterisation and subsequent targeted analysis (fatty acids, carbohydrates, and polyphenols). An Elute UHPLC 1300 coupled to a quadrupole time-of-flight mass spectrometry (QToF) Compact Instrument (Bruker Daltonics, Germany) was used. An Intensity Solo HPLC column C18 (2.0 μm, 100 mm × 2.1 mm) obtained from Bruker Daltonics was employed. The column temperature was set at 40 °C, and the injection volume was 2 μL. The mobile phases were constituted by A (water) and B (ethanol), both containing 0.1% of formic acid. The gradient starts at 95% (A) for 0.4 min, then increased up to 65% (A) in 0.1 min, and to 100% (B) in 7 min (held 5 min). The flow rate was 0.25 mL min−1, and the total run time was 15 min. The nebuliser pressure and temperature were set at 2 bar and 325 °C, respectively. The dry gas flow rate was set at 5 L min−1. Acquisition was done in the range m/z 10–1000 in negative and positive ionisation, showing the standards analysed in Supplementary Table S2.
For the identification of compounds by untargeted analysis, the computational package SmartFormula contained in the MetaboScape Version 4.0.4 (Build 19) analysis software was used. This tool provides the characterisation of analytes generated by fragmentation via UHPLC-QToF through comparison with the exact mass and isotopic profile of each compound. For the prediction of the molecular formula of the compounds, the compound crawler tool is used and corroborated by their structural stability with the MetFrag software, which combines the analysis of the in silico fragmentation of the possible candidates with the search in the main databases (ChemSpider, ChEBI, PubChem) of online chemical structures.
HPLC–DAD analysis (carotenoids)
For the quantification of the carotenoids present in the extracts, as the recovery study, a filtration process was carried out using a 0.22-μm PTFE membrane. Carotenoid standards were prepared in ethanol covering a concentration range with five levels (0.5, 1, 5, 10, 20 mg L−1). The same analytical rigour of the HPLC–DAD study was followed for the characterisation and quantification of the carotenoid compounds via UHPLC-QTOF. Coefficients of determination (R2) higher than 0.991 were obtained in all cases and are reported in Supplementary Figure S2. Quantification of the real samples was performed by external calibration using standard solutions of the studied compounds in ethanol, starting from a stock solution in DMF. Internal standard correction (apocarotenal) was used as an additional measure to control the procedure, but correction was not necessary.
A Jasco AS-4100 chromatographic equipment consisted of an AS-4150 autosampler coupled to a PU-4180 quaternary pump and an MD-4010 PDA detector was employed. The separation was conducted on a Kinetex C18 column (4.6 mm × 150 mm, 5 μm) (Phenomenex, USA). The column temperature was set at 30 °C, and the injection volume was 5 μL. The mobile phases were water (A) and methanol (B), both containing 1% of formic acid The flow rate was 1 mL min−1, starting with 80% of B, increasing progressively until 100% B in 10 min. The total run time was 25 min.
Determination of total polyphenolic index
The Folin-Ciocalteu assay was used to determine the total polyphenolic index of the extracts (Singleton and Rossi 1965). Briefly, a total of 5 mL of diluted extract (with a dilution factor of 40 in MiliQ water) was mixed with 100 μL of Folin-Ciocalteu reagent and shaken. Then, 1 mL of a concentrated solution of sodium carbonate 20% (w/v) prepared in ultrapure water was added. The solution was vortexed and isolated from light for 30 min. Then, the absorbance was measured at 760 nm. To express the total polyphenolic index values, gallic acid was used as calibration standard and reported as millimoles of gallic acid equivalent (mmolGAE) per gram of dry microalga (gDW).
Antioxidant activity assay
To evaluate the antioxidant activity of the samples, 2,2-diphenylyl-1-picrylhydrazyl radical (DPPH) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid (ABTS) assays were employed. For the DPPH assay, the guidelines described by Álvarez-Casas et al. (2016) were followed. Accordingly, 100 μL of diluted extract was introduced in a falcon tube, and 3.9 mL of 0.1 mM DPPH solution in methanol was added. The mixture was vortexed and isolated from light for 30 min. Then, the decrease in absorbance of the resulting mixture was measured at 515 nm. Quantification of the antioxidant activity was performed by comparison with the Trolox standard and expressed as Trolox Equivalent Antioxidant Activity (TEAC) per microalga dry weight as mmolTE g−1 DW. The uptake of ABTS radical cations (ABTS+) by the extracts was determined as described by Xiao et al. (2020) with minor modifications. Briefly, a 7 mM stock solution of ABTS was prepared in ethanol by reacting it with 2.45 mM potassium persulphate dissolved in ethanol. The mixture was stored in the dark at 25 °C for 16 h. The solution was dissolved in ethanol to obtain an absorbance of 0.700 (± 0.004) at 752 nm. The extracts were diluted to obtain a range of 10–80% inhibition when 50 μL of this was added to 4.5 mL of the stock solution. Extracts were allowed to react for 7 min with the ABTS+ radical in the dark. The result was expressed as mmolTE g−1 DW.
Results
Preliminary MSPD experiments
Solvent selection
Before MSPD optimisation, preliminary analysis to evaluate the influence of the elution solvent on the extraction efficiency of fatty acids, carotenoids, and carbohydrates was performed. Ethyl acetate (EtAc), ethyl lactate (lactate), and ethanol (EtOH) recognised by the Food and Drugs Administration (FDA) as GRAS solvents were tested and compared with acetone (AcOH), methanol (MeOH), and MTBE. Figure 2 illustrates the relative overall MSPD extractive profile for the three solvent groups considered from red stage H. pluvialis wet paste.

Overall MSPD extraction efficiency of three main families of compounds in aplanospore stage H. pluvialis wet paste (25% solid content) depending on the type of elution solvent used: GRAS (green scale), severely use-limited (AcOH, brown), and non-GRAS (MeOH and MTBE, black scale)
All tested solvents showed fairly similar results in terms of their carotenoid and fatty acid extraction efficiency. Amongst GRAS solvents, ethanol showed the highest extraction efficiency, only 4% lower than that of MTBE for carotenoids and 2% lower than that of methanol for fatty acids. To understand the behaviour of the different solvents tested, an analysis was then performed for each family of compounds: carotenoids, fatty acids, and carbohydrates. The solvent behaviour analysis for each family of compounds is detailed below.
Carotenoids
Once the overall carotenoid profile has been reported, an approach towards the individual response of these bioactive compounds is necessary. H. pluvialis in its red phase is characterised by a rich carotenoid content (up to 6%), with astaxanthin having the highest concentration (Cohen 1999). However, only 5% of the total astaxanthin is found in its free state, being generally present in esterified form, mainly coupled to palmitic, oleic, and linoleic acids (Shah et al. 2016). Figure 3a shows the individual response of the astaxanthin characterised in the preliminary study, as well as its respective monoesters (AME) coupled to C18:1 (oleic acid), C18:2 (linoleic acid), and C18:4 (stearidonic acid) acids for the selected solvents.

Relative response of the H. pluvialis paste main carotenoids and their esters in GRAS solvents (green scale), severely use-limited (AcOH, brown), and non-GRAS (MeOH and MTBE, black scale). AME astaxanthin monoester
In a second approach other carotenoids were identified in H. pluvialis paste: canthaxanthin, diadinoxanthin, and phoenicoxanthin, which extraction efficiency in the tested solvents is depicted in Fig. 3b. The response of the neoxanthin/violaxanthin and zeaxanthin/lutein isomers is plotted together.
Fatty acids
The relative response for the most relevant fatty acids in H. pluvialis wet paste was compared. Figure 4a shows the results obtained for the saturated fatty acids (SFA) and Fig. 4b those for the monounsaturated (MUFA) and polyunsaturated (PUFA) groups. The extraction efficiency presents a general trend: MeOH > EtOH > AcOH > MTBE > EtAc > lactate. To highlight the extractive behaviour of each solvent with respect to the overall mean of the respective compound, an average line is drawn.

Relative response of the H. pluvialis paste main fatty acids in GRAS solvents (green scale), severely use-limited (AcOH, brown), and non-GRAS (MeOH and MTBE, black scale)
Carbohydrates
Haematococcus pluvialis under stress conditions (e.g., temperature variations, nutrient starvation, light stress, and acidity) stored around 40% w/w of the total cellular carbohydrate content (Shah et al. 2016). To assess in detail the affinity profile of the solvents studied in the carbohydrate extraction, two main sugars, which represent the main carbohydrate content in the microalgae, are shown in Fig. 5. Hence, the high affinity for methanol, ethanol, and acetone is highlighted, in contrast to solvents such as ethyl lactate which generates an extract highly refined in these carbohydrate compounds.

Relative response of the H. pluvialis paste main carbohydrates in GRAS solvents (green scale), severely use-limited (AcOH, brown), and non-GRAS (MeOH and MTBE, black scale)
MSPD extraction optimisation design
The diverse extraction efficiency shown by the tested solvents for the fatty acids and carotenoids creates a compromise situation. It was necessary to select a solvent with the highest efficiency to extract the bioactive compounds being suitable for nutraceutical purposes. Compared to the non-GRAS solvents, ethanol showed an overall response 2% lower than the best solvent (methanol), presenting a uniform performance for the considered compounds, plus a stable profile within the analytes in each family. Thus, an optimisation procedure for extraction of wet red stage H. pluvialis via MSPD was carried out using ethanol as the extraction solvent.
To analyse the response of red stage concentrated paste (25% w/v) of H. pluvialis to the modification of the main extraction parameters by MSPD, an optimisation design was performed starting from the microalga in its wet state. In this fine design, the critical variables affecting extraction, sample mass, dispersant/sample ratio, and extraction solvent volume, were optimised. They were established based on previous experience in MSPD extractions using wet organic samples (Rubio et al. (2018) and Castillo et al. (2020)). Extraction (solvent elution) time and disruption time were set at 20 min and 10 min, respectively (Hoff and Pizzolato 2018). Table 1 summarises the factors and their selected levels.
In the case of the ratio dispersant/sample mass, a minimum value of 4 was established, due to the wet nature of the sample. Figure 6 shows that ratios lower than 4:1 resulted on a heterogeneous and agglutinated mixture, unsuitable for its treatment in the MSPD extraction column.

The MSPD column setup at dispersant:sample mass ratios 2:1 and 4:1
The main effect plots are depicted in the Pareto charts of Fig. 7, revealing statistical significance of the parameters A: sample size and C: extraction solvent volume, for both fatty acids and carotenoids.

Pareto charts showing the influence of A: sample size; B: dispersant/sample ratio; C: extraction solvent volume, on MSPD extraction of carotenoids and fatty acids from H. pluvialis wet paste
Although the dispersant:sample mass ratio does not show a statistically significant effect on the extraction of the compounds, a ratio 8:1 operationally improved both compaction and extractive bed size. However, a larger bed size generates a larger solvent path (higher tortuosity). This phenomenon is depicted in Fig. 8, which shows the effect of each parameter studied with the response obtained for carotenoids and fatty acids. In this last case, increasing the dispersant:sample mass ratio generated only a discrete improvement compared to its adverse effect on carotenoid extraction.

Effect of ethanol volume, sample size, and dispersant:sample mass ratio in the H. pluvialis wet paste (24% w/v) MSPD extraction
MSPD extraction efficiency
The optimised MSPD-based extraction process was able to generate a multicomponent extract from wet paste of H. pluvialis in its red stage without requiring any pre-treatment of the microalgae. The extraction efficiency of the proposed method was compared with reference methods for the isolation and recovery of carotenoids and fatty acids from microalgae. The results of the comparison, as well as the characterisation of the polyphenols and the evaluation of the antioxidant activity of the extract, are detailed below.
Carotenoids
The reference method (RM) selected to evaluate the efficiency of MSPD to extract carotenoids from wet H. pluvialis paste was that proposed by Kim et al. (2013) for the recovery of carotenoids from D. tertiolecta, adapted for the strong cell wall disruption of H. pluvialis. From Fig. 9, which provides a comparative graph of the extraction results achieved by both methods for free astaxanthin, canthaxanthin, beta-carotene, and the total of zeaxanthin/lutein isomers, it is clearly shown the improvement in the extraction of carotenoids when MSPD is applied. An average difference of 55% in the recovery between both methods has been obtained, showing a similar behaviour for all compounds. This better efficiency of MSPD for carotenoid extraction was also found in our previous study using lyophilised H. pluvialis (Castillo et al. 2020).

Comparison of the extraction efficiency of MSPD vs reference method (RM) for the main carotenoids in red stage H. pluvialis wet paste (25% w/v)
Fatty acids
The extraction efficiency of fatty acids by MSPD was compared to the Bligh and Dyer (1959) RM. This method has been tested in H. pluvialis for different lipid chains (from C12 to C20) extraction, generating the highest global results compared to methodologies such as Folch, hypochlorous acid treatment, and acid-catalysed hot-water extraction (Otero et al. 2017). Besides, the option of a prior saponification of H. pluvialis was discarded, as its effect has been proved to be indifferent or disadvantageous on the overall lipid recovery (Otero et al. 2017). The result of the comparative analysis is shown in Fig. 10.

Comparison of the extraction efficiency of the MSPD vs reference method (RM) for the main fatty acids contained in red stage H. pluvialis wet paste (25% w/v)
The fatty acids were divided into 2 groups according to the concentration in the microalgae. In addition, a blue-coloured surface is plotted to highlight the absolute difference of the recoveries obtained by both methods. This surface reveals a higher relative extraction by the MSPD as a function of the RM when the unsaturation in the lipid compounds increases. In the case of palmitic acid (C16:0), the recovery by RM is 15% lower, decreasing slightly for linoleic acid (C18:2) with 22%. However, for linolenic acid (C18:3), the difference is even more marked, with MSPD showing a 68% improvement in extraction.
Polyphenolic comprehensive analysis
In a first approach, the total polyphenol content (TPC) of the extract was evaluated by the Folin-Ciocalteu method, obtaining a total of 24.65 mgGAE g−1 DW for the MSPD ethanol extract of the wet paste red stage H. pluvialis. A remarkable aspect of the phenolic content of H. pluvialis is that, although a proportionality with the concentration of carotenoids is known, there is limited information regarding the phenolic compounds responsible for the bioactivities of the microalga (Deniz 2020). Thus, to uncover the polyphenolic profile of H. pluvialis, an untargeted analysis of the extract was performed, aimed to the characterisation of polyphenols.
The criteria used for the untargeted identification and confirmation of the compounds follows a precise validation algorithm previously used for the analysis of H. pluvialis (Castillo et al. 2020), as well as extracts of natural origin via ESI-QToF (Ríos et al. 2015). The process starts with the analysis of the analyte intensity, which must be above the confidence limit of the instrument. If approved, the candidate must meet two main parameters: its isotopic standard and mass deviation. The isotopic pattern, reflected as mSigma, shows the effective ratio of the experimental mass fragmentation to the theoretical basis. Likewise, the mass deviation expresses the deviation between the detected experimental mass and the theoretical mass. As upper limit values, 50mSigma and 5mDa are set, which generates an efficient screening that is then supported by the targeted analysis. In this way, applying the algorithm described above, the phenolic compounds phloroglucinol, p-coumaric acid, gallic acid, and catechin, were identified (Table 2).
Due to their low concentration in the extract, catechin and gallic acid presented a profile that did not allow mSigma values below the established limit, and their identification was initially considered as tentative. In a subsequent targeted analysis, the phenolics compounds (phloroglucinol, p-coumaric acid, gallic acid, and catechin) were identified and quantified by comparison with their respective standards.
Phloroglucinol and p-coumaric acid stand out amongst the reduced number of studies dealing with the polyphenolic profile of H. pluvialis. These compounds have not been previously reported in red stage of H. pluvialis, although a study by Goiris et al. (2014) focused on the search for different compounds of the family of flavonoids, cinnamic acids and simple phenols, revealed that phloroglucinol represented around 99% (81 μg g−1) of the total phenolic compounds in the green phase of the microalga, with more than 70% (0.6 μg g−1) of the remaining amount accounted by p-coumaric acid. The results found for the red stage paste show a similar profile (Fig. 11), although the phloroglucinol concentration was higher (2.3 mg g−1), with 32.0 μg g−1 of p-coumaric acid, and much lower of catechin (10.0 μg g−1) and gallic acid (7.8 μg g−1).

Polyphenols relative concentrations found in the paste of red stage H. pluvialis
Antioxidant activity
For the antioxidant activity evaluation of MSPD wet paste H. pluvialis extracts, the DPPH analysis was initially performed. It is common to quantify the antioxidant activity of carotenoids using the DPPH radical at a maximum wavelength of 515 nm. However, only one study reports the possible interference between the absorbance of carotenoids contained in H. pluvialis (400–500 nm) with the DPPH reagent (Santoyo et al. 2010). Several dilutions of the H. pluvialis extract in methanol were carried out to check such interference. As shown in supplementary Fig. S3, and considering that absorbance is an additive property, from a dilution factor range from 140 up to 500 (v/v), a marked interference is observed at 515 nm in the absorption of the extracts related to that of DPPH radical. From a dilution factor of 1000, a measurement without significant interference is obtained, but at this dilution the extract is unable to give an appreciable response in the free radical inhibition. Hence, despite the common practice of performing the DPPH procedure to measure the antioxidant activity of carotenoids, and due to the proven impossibility of measuring the antioxidant activity with this methodology, the ABTS radical method was alternatively used. ABTS cation has its maximum absorption at 645, 734, and 815 nm when ethanol is used as the working solvent, which does not interfere with the absorption of the sample analysed (Re et al. 1999). Accordingly, the antioxidant value obtained for red stage H. pluvialis wet paste by the proposed MSPD extraction method was 1.58 mmolTE g−1 DW.
Discussion
There are several proposals seeking an answer to the choice of the appropriate solvent for the extraction of bioactive compounds from microalgae. MTBE has been a widely used solvent for the extraction of carotenoid compounds from microalgae (Mojaat et al. 2008). Due to its high affinity for carotenoids and high volatility, which allows for rapid removal and recovery, it is a common solvent for astaxanthin recovery studies (Bauer and Minceva 2019). Solvents such as methanol and acetone are also commonly used for the extraction of astaxanthin from H. pluvialis (Molino et al. 2018; Park et al. 2020).
Regarding the application of GRAS as extraction solvents in H. pluvialis, only one previous work showed the application of ethyl lactate (Castillo et al. 2020), whilst there are few studies in which this solvent is applied for the extraction of microalgal compounds for nutritional purposes. In the case of Scenedesmus obliquus, comparable results have been obtained for fatty acid recovery and antioxidant activity value using ethyl lactate and solvents such as acetone, isopropanol, and hexane, (Amaro et al. 2015). Lactate was also used with Chlorella vulgaris and Nannochloropsis sp. showing considerable affinity for saturated and monounsaturated fatty acids, as opposed to polyunsaturated fatty acids. (Wan et al. 2017). Ethyl acetate and ethanol have been widely used in the microalgae field, with ethanol being the GRAS solvent that has shown the highest extractive efficiency for most of the bioactive compounds of interest in H. pluvialis (Irshad, et al. 2019b). In this way, although all the solvents chosen in this study present several advantages in the extraction of bioactive compounds from microalgae, making a direct comparison is somewhat complex, but undoubtedly interesting. As summarised in the optimization of the proposed MSPD extraction procedure in this work, a similar affinity to the selected solvents is observed for SFA C18:1 (oleic acid), C18:2 (linoleic acid), and C18:4 (stearidonic acid). The increased response when MeOH is used as an extractant for SFA in contrast to free astaxanthin is notable. The astaxanthin extraction efficiency is dependent on the nature of the solvent, resulting in the following trend for this assay: methanol > ethanol > acetone. This behaviour was previously reported by Haque et al. (2016) and Molino et al. (2018), who detailed the effects of solvents on astaxanthin recovery by ultrasonication and accelerated solvent extraction (ASE), respectively. Canthaxanthin and phoenicoxanthin show a similar profile. Diadinoxanthin is better extracted by acetone, and the zeaxanthin/lutein isomers by ethanol. For the neoxanthin/violaxanthin isomers, all the solvents tested presented similar extraction efficiency.
Methanol and ethanol showed a stable extraction profile towards the fatty acids, as opposed to ethyl acetate, acetone, and MTBE which showed lower extraction efficiency with the length of the aliphatic chain. For ethyl lactate, a more defined selectivity is observed which is altered by other factors than lipid length. For the SFA and MUFA groups, the response obtained using ethyl lactate as solvent was 75% of the overall mean, in contrast to PUFA, for which this value drops to only 5%. PUFAS are of special interest from a nutritional point of view, as they include omega-3 and omega-6 compounds, useful in the prevention of cardiovascular diseases (Artemis 2008). In this way, a two-step extractive process, where SFA and MUFA were extracted in a first step, would be promising to obtain a final extract mainly loaded with these PUFA.
Haematococcus pluvialis in the red stage can store up to 40% carbohydrates (Shah et al. 2016). Most of these carbohydrates are sugars such as glucose and sucrose, which are hydrophilic in nature, displaying a better response of these compounds towards solvents with upper polarity (methanol > ethanol > acetone). In contrast, it is remarkable that ethyl lactate does not extract sugars, showing that not only polarity affects the extraction selectivity, as it is comparable to that of ethanol, but allows the obtention of a sugar-free extract when desired. Current polysaccharide refining methods involve hydrolysis techniques using conditions that are not compatible with a nutraceutical purpose, such as acidic, alkaline media, or physical methods using high temperatures, MAE, and UAE (Suarez et al. 2020). These processes involve demanding energy requirements coupled with a complex scale-up. Thus, the selective extraction by the solvents ethyl lactate and ethyl acetate creates an attractive GRAS alternative for the removal of carbohydrate content, whilst keeping the lipid and carotenoid profile comparable to the other solvents.
Due to the compromise generated by the heterogeneous extractive profile described, a decisive choice of working solvent was essential. In line with a nutraceutical approach in combination with an overall positive overall response of the solvents tested, the choice of a GRAS extractant was clear, where ethanol was outstanding. On the one hand, it gave the best overall response within its category, and on the other hand it has no restrictions, beyond those governing good manufacturing practices, for its application in food formulations, unlike acetone, which is considered within this category when its use is less than 5 to 8 mg L−1 (Molino et al. 2018).
The efficiency and selectivity of the MSPD extraction depend on numerous factors, including the physical state and origin of the sample, the relative concentrations and properties of the compounds, and an appropriate combination of dispersant and elution solvent (Basheer et al. 2012). It is also remarkable for its ability to sometimes integrate extraction and clean-up in the same process (García-López et al. 2008). The RM applied in this work in the recovery of carotenoids implied multiple and extensive solvent washes, liquid–liquid separation with subsequent volatilisation, and reconstitution. These subsequent steps aimed at a carotenoid-refined extract could increase experimental error and possible loss of material. In contrast, the proposed MSPD procedure combines extraction, maceration, and filtration in a single step, without the need for reconstitution since the working solvent is appropriate for the straightforward liquid chromatographic analysis.
It could be assumed that lipids of the same group, differentiated only by an unsaturation level, such as linoleic acid and linolenic acid, would present a constant relative response between the two methodologies analysed. Hence, the considerable extractive improvement of MSPD compared with the RM for linolenic acid and linoleic acid is striking. However, depending on the polarity and affinity of the fatty acid in question, the extraction efficiency could differ. An example of this is evidenced by Otero et al. (2017) when comparing the method of Bligh and Dyer (1959) and Folch. When extracting linolenic acid contained in H. pluvialis, a similar profile is observed between both methods, but not for linoleic acid, which is much better extracted by the proposed MSPD method. As for the minority saturated fatty acids (Fig. 10B), MSPD also shows an improved efficiency compared to the RM.
Given the antioxidant properties of phenolic compounds (Cory et al. 2018) and the potential of microalgae as sources of these compounds (Del Mondo et al. 2021), few studies have addressed their identification and quantification in microalgae matrices (Jerez-Martel et al. 2017). Consequently, the value of 24.65 mgGAE g−1 DW obtained by Folin-Ciocalteu assay in this study for the MSPD extract of red stage H. pluvialis paste is not a simple measure to compare. The range of total phenolic content (TPC) reported for H. pluvialis, although limited, is heterogeneous, as it is highly dependent on the different extractive procedures, solvents, and stage of the microalga. A study conducted for the red and green stages of H. pluvialis shows values of 0.54 and 1.23 mgGAE g−1 DW, respectively (Goiris et al. 2012). These values, much lower than those reported in this work, were obtained by mortar disruption with no apparent disruptive medium. Since the cell wall of H. pluvialis is characterised by its rigidity (Ye et al. 2020), a precarious disruption in the absence of disruptive material can be assumed. Additionality, proportions of ethanol diluted in a volume of water were used, which made cell wall degradation even more difficult. Other extractive procedure, through the generation of oleoresin, gave phenolic index values of about 75mgGAE g(oleoresin)−1 (Serwotka-Suszczak et al. 2019). This value of TPC represents the highest index ever reported for H. pluvialis. However, it should be considered that the result is a semi-solid extract of considerable concentration, expressing its activity in mass of oleoresin produced, thus avoiding direct comparison with the ethanolic MSPD extract. In other microalgae species such as Spirulina platensis and Chlorella pyrenoidosa, values between 24.4 and 25.8 mgGAE g−1 DW were obtained, using 100% methanol as the extractive solvent (Machu et al. 2015). These values are comparable to those obtained in the present study.
Regarding the analysis of phenolic compounds, there is a somewhat widespread belief in the inability of microalgae to synthesise flavonoids. The identification of these compounds in microalgae matrices is relatively recent, and the range of data available to be compared with the obtained in this study is not very wide. Jerez-Martel et al. (2017) report in microalgae species such as Euglena cantabrica and Spirogyra sp. as in the cyanobacterium Nostoc commune; different values of gallic acid correspond to 5.8 μg g−1, 91.4 μg g−1, and 71 μg g−1; respectively. Only E. cantabrica shows catechin concentrations about 71.4 μg g−1 (Jerez-Martel et al. 2017).
Through the in-depth characterisation of H. pluvialis in the red aplanospore stage extracts obtained by an optimised procedure of extraction based on MSPD, it can be concluded that the bioactive power of the microalgae, although strongly linked to its high concentration of compounds such as carotenoids and fatty acids, has a relevant impact on the synergistic action of phenolic compounds. In this way, it has been reported that the unique effect of phloroglucinol, the most abundant microalgae polyphenol, in several algal species is able to provide between 1 and 10% of the total response when bioactivities such as antioxidant capacity are measured (Goiris et al. 2012; Quéguineur et al. 2012). However, contrasting this value with those obtained by current methodologies is a challenge. In general, the measurement of antioxidant activity is performed on purified astaxanthin from H. pluvialis, but not on the microalgal extract. Extracts from lyophilised red stage H. pluvialis obtained by CO2-expanded ethanol extraction (Reyes et al. 2014) and pressurised liquid extraction (PLE) (Jaime et al. 2010) show antioxidant activity values ranging from 0.084 to 0.267 mmolTE g(extract)−1 respectively. These procedures, being referred to the extract mass, make comparison with the values obtained in the present work even more difficult. If the density of the ethanolic extract obtained by MPSD is assumed to be that of the pure solvent, the antioxidant activity value derived would be 0.040 mmolTE g(extract)−1. This value, although lower than those referenced above, is comparable to those obtained by very strong extractive processes, such as those combining freezing with liquid N2, and solvents such as hexane at temperatures in the range of 50–200 °C with pressures above 10 MPa (Reyes et al. 2014) (Jaime et al. 2010). These conditions make it possible to obtain a more concentrated extract by modifying the capacity of the solvents used to solubilise the carotenoid compounds. On the contrary, if the extractive capacity is evaluated considering the starting mass of the microalgae, MSPD generates antioxidant activity values equal to or higher than those obtained by these methodologies.
Another common way to evaluate the antioxidant power generated mainly by the natural astaxanthin contained in H. pluvialis is by contrast with its synthetic alternative. Thus, the mean TEAC values of synthetic astaxanthin range between 2.21 and 2.43 mmolTE g−1 DW (Jaime et al. 2010; Régnier et al. 2015) which, despite being higher than the values obtained for the ethanol extract of H. pluvialis MSPD in red phase, is synthesised by petrochemical processes that often raise alarming food safety issues with potential toxicity in the final product (Jeevanandam et al. 2020). In addition, synthetic production also presents challenges in terms of sustainability and potential environmental impact in the manufacturing process (Li et al. 2011). These drawbacks have confined the use of this synthetic palliative to the fish feed additive sector for organoleptic (pigmentation) purposes, and it is not approved for human consumption or as a food additive (Capelli et al. 2013).
Conclusions
The unidirectional approach to the refining of astaxanthin from H. pluvialis should be transformed into a whole in order to make it feasible. An optimal extract could be obtained from the wet microalga via MSPD, not needing previous lyophilisation of the substrate, showing the best recoveries, as well as bioactive power when ethanol is used as the extraction solvent. In turn, ethyl lactate as a GRAS alternative, gave extracts with an interesting lipid extraction profile of the form SFA > MUFA > PUFA and no carbohydrate content. The ethanolic extract, which has no limitations in food use, showed an antioxidant power of 1.58 mmolTE g−1 DW, comparable to its synthetic pure astaxanthin counterpart. In-depth characterisation of the MSPD extract composition revealed that the synergistic activity of minor polyphenols, in combination with significant concentrations of phloroglucinol (2.3 mg g-1), has a direct effect on the total phenolic content of H. pluvialis (24.65 mgGAE g−1 DW). Therefore, the combination of GRAS solvents with a scalable MSPD method, consistent with a nutraceutical purpose, is a very suitable, green, and low-cost alternative not only to obtain astaxanthin, but also to extract a high number of bioactive compounds from H. pluvialis.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary material files.
References
Ahmed F, Li Y, Fanning K, Netzel M, Schenk PM (2015) Effect of drying, storage temperature and air exposure on astaxanthin stability from Haematococcus pluvialis. Food Res Int 74:231–236
Álvarez-Casas M, Pájaro M, Lores M, Garcia-Jares C (2016) Characterization of grape marcs from native and foreign white varieties grown in northwestern Spain by their polyphenolic composition and antioxidant activity. Eur Food Res Technol 242:655–665
Amaro H, Fernandes F, Valentão P, Andrade P, Sousa-Pinto I, Malcata F, Guedes A (2015) Effect of solvent system on extractability of lipidic components of Scenedesmus obliquus (M2–1) and Gloeothece sp. on antioxidant scavenging capacity thereof. Mar Drugs 13:6453–6471
Anastas PT, Warner JC (1998) Green chemistry: theory and practice. Oxford University Press, Oxford
Artemis PS (2008) The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med 233:674–688
Barker SA, Long AR, Short CR (1989) Isolation of drug residues from tissues by solid phase dispersion. J Chromatogr A 475:353–361
Basheer C, Alhooshani K, Nuhu AA, Kanimozhi S, Lee HK (2012) Sample preparation of complex biological samples in the analysis of trace-level contaminants. Compr Samp, Samp Prep 3:681–700
Bauer A, Minceva M (2019) Direct extraction of astaxanthin from the microalgae: Haematococcus pluvialis using liquid-liquid chromatography. RSC Adv 9:22779–22789
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917
Capelli B, Bagchi D, Cysewski GR (2013) Synthetic astaxanthin is significantly inferior to algal-based astaxanthin as an antioxidant and may not be suitable as a human nutraceutical supplement. Nutrafoods 12:145–152
Castillo A, Pereira S, Otero A, Fiol S, Garcia-Jares C, Lores M (2020) Matrix solid-phase dispersion as a greener alternative to obtain bioactive extracts from: Haematococcus pluvialis. characterization by UHPLC-QToF. RSC Adv 10:27995–28006
Cerón MC, García-Malea MC, Rivas J, Acien FG, Fernandez JM, Del Río E, Guerrero MG, Molina E (2007) Antioxidant activity of Haematococcus pluvialis cells grown in continuous culture as a function of their carotenoid and fatty acid content. Appl Microbiol Biotechnol 74:1112–1119
Chen W, Liu Y, Song L, Sommerfeld M, Hu Q (2020) Automated accelerated solvent extraction method for total lipid analysis of microalgae. Algal Res 51:102080
Cohen Z (1999) Chemicals from microalgae. CRC Press, Boca Raton
Cory H, Passarelli S, Szeto J, Tamez M, Mattei J (2018) The role of polyphenols in human health and food systems: a mini-review. Front Nutr 5:87
Del Mondo A, Smerilli A, Ambrosino L, Albin A, Noonan DM, Sansone C, Brunet C (2021) Insights into phenolic compounds from microalgae: structural variety and complex beneficial activities from health to nutraceutics. Crit Rev Biotechnol 41:155–171
Deniz I (2020) Scaling-up of Haematococcus pluvialis production in stirred tank photobioreactor. Bioresour Technol 310:123434
Do TT, Ong BN, Tran M-LN, Nguyen D, Melkonian M, Tran H (2016) D. Biomass and astaxanthin productivities of Haematococcus pluvialis in an angled twin-layer porous substrate photobioreactor: effect of inoculum density and storage time. Biology 8:68
Fábregas J, Domínguez A, Regueiro M, Maseda A, Otero A (2000) Optimisation of culture medium for the continuous cultivation of the microalga Haematococcus pluvialis. Appl Microbiol Biotechnol 53:530–535
García-López M, Canosa P, Rodríguez I (2008) Trends and recent applications of matrix solid-phase dispersion. Anal Bioanal Chem 391:963–974
Gato E, Perez A, Rosalowska A, Celeiro M, Bou G, Lores M (2021) Multicomponent polyphenolic extracts from Vaccinium corymbosum at lab and pilot scale. Characterization and effectivity against nosocomial pathogens. Plants 10:2801
Goiris K, Muylaert K, Fraeye I, Foubert I, de Brabanter J, de Cooman L (2012) Antioxidant potential of microalgae in relation to their phenolic and carotenoid content. J Appl Phycol 24:1477–1486
Goiris K, Muylaert K, Voorspoels S, Noten B, De Paepe D, Baart GJ, De Cooman L (2014) Detection of flavonoids in microalgae from different evolutionary lineages. J Phycol 50:483–492
Haque F, Dutta A, Thimmanagari M, Chiang YW (2016) Intensified green production of astaxanthin from Haematococcus pluvialis. Food Bioprod Process 99:1–11
Hoff RB, Pizzolato TM (2018) Combining extraction and purification steps in sample preparation for environmental matrices: a review of matrix solid phase dispersion (MSPD) and pressurized liquid extraction (PLE) applications. Trends Anal Chem 109:83–96
Huang W-C, Liu H, Sun W, Xue C, Mao X (2018) Effective astaxanthin extraction from wet Haematococcus pluvialis using switchable hydrophilicity solvents. ACS Sustain Chem Eng 6:1560–1563
Irshad M, Hong ME, Myint AA, Kim J, Sim SJ (2019a) Safe and complete extraction of astaxanthin from Haematococcus pluvialis by efficient mechanical disruption of cyst cell wall. Int J Food Eng 15:20190128
Irshad M, Myint AA, Hong ME, Kim J, Sim SJ (2019b) One-pot simultaneous cell wall disruption and complete extraction of astaxanthin from Haematococcus pluvialis at room temperature. ACS Sustain Chem Eng 7:13898–13910
Jaime L, Rodríguez-Meizoso I, Cifuentes A, Santoyo S, Suarez S, Ibáñez E, Señorans FJ (2010) Pressurized liquids as an alternative process to antioxidant carotenoids extraction from Haematococcus pluvialis microalgae. LWT Food Sci Technol 43:105–112
Jannel S, Caro Y, Bermudes M, Petit T (2020) Novel insights into the biotechnological production of Haematococcus pluvialis-derived astaxanthin: advances and key challenges to allow its industrial use as novel food ingredient. J Mar Sci 8:789
Jeevanandam J, Choudhary V, Selvam JD, Danquah MK (2020) The bioeconomy of production of microalgal pigments. In: Jacob-Lopes E, Queiroz M, Zepka L (eds) Pigments from microalgae handbook. Springer, Cham, pp 325–363
Jerez-Martel I, García-Poza S, Rodríguez-Martel G, Rico M, Afonso-Olivares C, Gómez-Pinchetti JL (2017) Phenolic profile and antioxidant activity of crude extracts from microalgae and cyanobacteria strains. J Food Qual 2017:2924508
Jin G, Liu Y, Xue S, Meng Y, Yan J, Yang F, Guo Z, Zhu J, Liang X (2019) Determination of three carotenoids in microalgae by matrix solid-phase dispersion extraction and high-performance liquid chromatography. Chromatographia 82:1593–1601
Kapoore R, Butler T, Pandhal J, Vaidyanathan S (2018) Microwave-assisted extraction for microalgae: from biofuels to biorefinery. Biology 7:18
Kim SH, Liu KH, Lee SY, Hong SJ, Cho BK, Lee H, Lee CG, Choi HK (2013) Effects of light intensity and nitrogen starvation on glycerolipid, glycerophospholipid, and carotenoid composition in Dunaliella tertiolecta culture. PLoS ONE 8:72415
Li J, Zhu D, Niu J, Shen S, Wang G (2011) An economic assessment of astaxanthin production by large scale cultivation of Haematococcus pluvialis. Biotechnol Adv 29:568–574
Lorenz RT, Cysewski GR (2000) Commercial potential for Haematococcus microalgae as a natural source of astaxanthin. Trends Biotechnol 18:160–167
Lores M, Alvarez-Casas M, Garcia-Jares C, Llompart M (2016) Polyphenol extract from white-grape residue. European Patent EP2875822A4
Machu L, Misurcova L, Ambrozova JV, Orsavova J, Mlcek J, Sochor J, Jurikova T (2015) Phenolic content and antioxidant capacity in algal food products. Mol 20:1118–1133
Mojaat M, Foucault A, Pruvost J, Legrand J (2008) Optimal selection of organic solvents for biocompatible extraction of β-carotene from Dunaliella salina. J Biotechnol 133:433–441
Molina E, Belarbi EH, Fernández FG, Medina A, Chisti Y (2003) Recovery of microalgal biomass and metabolites: process options and economics. Biotechnol Adv 20:491–515
Molino A, Rimauro J, Casella P, Cerbone A, Larocca V, Chianese S, Karatza D, Mehariya S, Ferraro A, Hristoforou E, Musmarra D (2018) Extraction of astaxanthin from microalga Haematococcus pluvialis in red phase by using generally recognised as safe solvents and accelerated extraction. J Biotechnol 283:51–61
Mularczyk M, Michalak I, Marycz K (2020) Astaxanthin and other nutrients from Haematococcus pluvialis—multifunctional applications. Mar Drugs 18:459
Otero P, Saha SK, Gushin JM, Moane S, Barron J, Murray P (2017) Identification of optimum fatty acid extraction methods for two different microalgae Phaeodactylum tricornutum and Haematococcus pluvialis for food and biodiesel applications. Anal Bioanal Chem 409:4659–4667
Panis G, Carreon JR (2016) Commercial astaxanthin production derived by green alga Haematococcus pluvialis: a microalgae process model and a techno-economic assessment all through production line. Algal Res 18:175–190
Park JY, Oh YK, Choi SA, Kim MC (2020) Recovery of astaxanthin-containing oil from Haematococcus pluvialis by nano-dispersion and oil partitioning. Appl Biochem Biotechnol 190:1304–1318
Pereira S, Otero A (2020) Haematococcus pluvialis bioprocess optimisation: effect of light quality, temperature and irradiance on growth, pigment content and photosynthetic response. Algal Res 51:102027
Quéguineur B, Goya L, Ramos S, Martín MA, Mateos R, Bravo L (2012) Phloroglucinol: antioxidant properties and effects on cellular oxidative markers in human HepG2 cell line. Food Chem Toxicol 50:2886–2893
Rama LR, Mallo N, Biddau M, Fernandes F, de Miguel T, Sheiner L, Choupina A, Lores M (2021) Exploring the powerful phytoarsenal of white grape marc against bacteria and parasites causing significant diseases. Environ Sci Pollut Res Int 28:24270–24278
Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C (1999) Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med 26:1231–1237
Régnier P, Bastias J, Rodriguez-Ruiz V, Caballero-Casero N, Caballo C, Sicilia D, Fuentes A, Maire M, Crepin M, Letourneur D, Gueguen V, Rubio S, Pavon-Djavid G (2015) Astaxanthin from Haematococcus pluvialis prevents oxidative stress on human endothelial cells without toxicity. Mar Drugs 13:2857–2874
Reyes FA, Mendiola JA, Ibañez E, del Valle JM (2014) Astaxanthin extraction from Haematococcus pluvialis using CO2-expanded ethanol. J Supercrit Fluids 92:75–83
Ríos JJ, Roca M, Pérez-Gálvez A (2015) Systematic HPLC/ESI-high resolution-QTOF-MS methodology for metabolomic studies in nonfluorescent chlorophyll catabolites pathway. J Anal Methods Chem 2015:1–10
Rubio L, Lamas JP, Lores M, Garcia-Jares C (2018) Matrix solid-phase dispersion using limonene as greener alternative for grape seeds extraction, followed by GC-MS analysis for varietal fatty acid profiling. Food Anal Methods 11:3235–3242
Ruen-ngam D, Shotipruk A, Pavasant P (2011) Comparison of extraction methods for recovery of astaxanthin from Haematococcus pluvialis. Sep Sci Technol 46:64–70
Santoyo S, Plaza M, Jaime L, Ibañez E, Reglero G, Señorans FJ (2010) Pressurized liquid extraction as an alternative process to obtain antiviral agents from the edible microalga Chlorella vulgaris. J Agric Food Chem 58:8522–8527
Serwotka-Suszczak AM, Marcinkowska KA, Smieszek A, Michalak IM, Grzebyk M, Wiśniewski M, Marycz KM (2019) The Haematococcus pluvialis extract enriched by bioaccumulation process with mg (ii) ions improves insulin resistance in equine adipose-derived stromal cells (EqASCs). Biomed Pharmacother 116:108972
Shah MR, Liang Y, Cheng JJ, Daroch M (2016) Astaxanthin-producing green microalga Haematococcus pluvialis: from single cell to high value commercial products. Front Plant Sci 7:531
Singleton VL, Rossi JA (1965) Colorimetry of total phenolics with phosphomolybdic-phosphotungstic acid reagents. Am J Enol Vitic 16:144–158
Stramarkou M, Papadaki S, Kyriakopoulou K, Krokida M (2017) Effect of drying and extraction conditions on the recovery of bioactive compounds from Chlorella vulgaris. J Appl Phycol 29:2947–2960
Suarez CA, Baca SZ, van den Broek LAM, van den Berg C, Wijffels RH, Eppink MHM (2020) Selective fractionation of free glucose and starch from microalgae using aqueous two-phase systems. Algal Res 46:101801
Wan WMA, Theodoropoulos C, Gonzalez-Miquel M (2017) Enhanced microalgal lipid extraction using bio-based solvents for sustainable biofuel production. Green Chem 19:5723–5733
Xiao F, Xu T, Lu B, Liu R (2020) Guidelines for antioxidant assays for food components. Food Front 1:60–69
Ye Z, Tan XH, Liu ZW, Aadil RM, Tan YC, Inam-ur-Raheem M (2020) Mechanisms of breakdown of Haematococcus pluvialis cell wall by ionic liquids, hydrochloric acid and multi-enzyme treatment. Int J Food Sci Technol 55:3182–3189
Zhao L, Zhao G, Chen F, Wang Z, Wu J, Hu X (2006) different effects of microwave and ultrasound on the stability of (all-E)-astaxanthin. J Agric Food Chem 54:8346–8351
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This research was funded by project ED431 2020/06 (Galician Competitive Research Groups Xunta de Galicia). This work was supported by project EQC2018-005011-P (Ministry of Science, Innovation and Universities, Spain). All these programmes are co-funded by FEDER (EU). This article is based upon work from the Sample Preparation Study Group and Network, supported by the Division of Analytical Chemistry of the European Chemical Society.
Author information
Authors and Affiliations
Contributions
Conceptualization, A. O., C. G.-J., and M. L.; methodology, A. O., S. P., C. G.-J., and M. L.; formal analysis, C. G.-J. and M. L.; investigation, A. C., S. P.; writing—original draft preparation, A. C.; writing—review and editing, C. G.-J., and M. L.; supervision, C. G.-J. and M. L.; project administration, C. G.-J. and M. L.; funding acquisition, A. O, M. L
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Castillo, A., Pereira, S., Otero, A. et al. Multicomponent bioactive extract from red stage Haematococcus pluvialis wet paste: avoiding the drying step and toxic solvents. J Appl Phycol 34, 1537–1553 (2022). https://doi.org/10.1007/s10811-022-02712-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-022-02712-3